Short overview
Polar bears are a powerful symbol of the strength and endurance of the Arctic. Habitat fragmentation and contraction caused by human activities in the past century pose a serious threat to the genetic diversity of many species, including polar bears, and increase their risk of extinction. The aim of this study is to investigate the genetic diversity, population structure, migration trends, and demographic history of polar bear populations in the Russian Arctic.


Extended project overview
The polar bear is the world’s largest terrestrial carnivore and a dominant predator in the Arctic, roaming the sea ice and tundra. This charismatic species is the only land mammal that uses pack ice as its main habitat. The polar bear’s Latin name, Ursus maritimus, means “sea bear.”. These bears are strong swimmers and can cover vast distances in icy waters.


Polar bears are distributed in 20 relatively distinct populations across the circumpolar Arctic, ranging in size from a few hundred to a few thousand individuals. In the Russian Arctic, there are three subpopulations: the Kara-Barents, Laptev, and Chukotka-Alaska populations. However, the genetic status of these subpopulations is largely unknown, which limits our conservation efforts and ability to protect polar bears.
The past century has seen an increase in human activity and infrastructure development in the Russian Arctic, leading to animal habitat fragmentation and contraction. This poses a significant threat to the genetic diversity of many species, including polar bears, and increases their risk of extinction.



To address these issues, this project aims to uncover the genetic diversity, population structure, and demographic history of polar bear subpopulations in the Russian Arctic. Specifically, the study seeks to answer the question: “How has the genetic status of polar bear subpopulations changed since the 1900s?”

By providing this information, we can better understand the threats facing polar bears and take more effective action to protect their well-being.
 © Hans-Jurgen Mager
© Hans-Jurgen MagerThis project aims to provide a comprehensive understanding of the genetic variability, population structure, and demographic history of polar bears in the Russian Arctic. By sequencing archival and modern DNA samples collected from various habitats in the region, including samples dating back to the 1880s, we aim to produce a detailed description of the polar bear population in the Russian Arctic. The project’s results have the potential to fill key gaps in our knowledge of polar bear population genetics and help us understand the impact of human activity on polar bear populations in the past century. Additionally, the findings will facilitate the development of molecular genetic tools for preventing poaching and monitoring migrations.
Overall, this project is an essential step towards protecting the well-being of polar bears and preserving the fragile Arctic ecosystem in which they live.
 © Dan Bolton
© Dan Bolton



2022
September
By the shores of Neva River, by the shining Finnish Gulf in the very heart of Saint Petersburg stands the Zoological Institute of the Russian Academy of Sciences (https://www.zin.ru).


The Zoological Institute of the Russian Academy of Sciences (https://www.zin.ru) in Saint Petersburg is a treasure trove for zoology enthusiasts and researchers. The Institute houses one of the largest and most diverse zoological collections in the world, with approximately 60 million items that represent roughly 25% of the world’s fauna, spanning over 260 different species.



Among these remarkable specimens is the polar bear collection, which includes items dating back as far as the heroic age of Polar exploration in the mid-1800s. However, the bulk of the collection is from the 1930s-1940s, a time when the entire Russian North, from the Pechora to Chukotka, was actively explored under the Northern Sea Route development plan.


The catalogue of the collection is a reflection of the era, with specimens donated to the Institute by famous Polar explorers such as Alexander von Bunge, Leonid Starokadomsky, Boris Vilkitsky, and others.



We are grateful for the privilege of working within the walls of this remarkable institution every day for the next couple of months.
2022
October-November
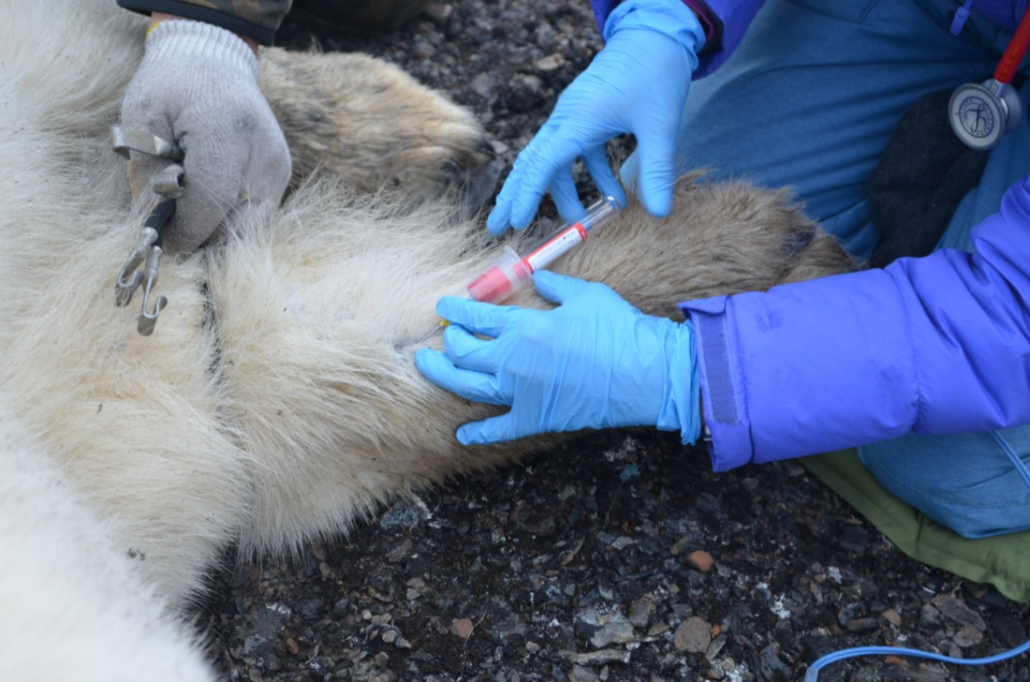
The Institute’s polar bear collection mainly consists of complete bear skulls, although in certain cases, the rest of the skeleton and skins are also available.



Skeletal remains and sediments can preserve DNA for hundreds of thousands of years, and recent studies have shown that ancient/historical DNA is best preserved in cranial base (petrous bone) or teeth.

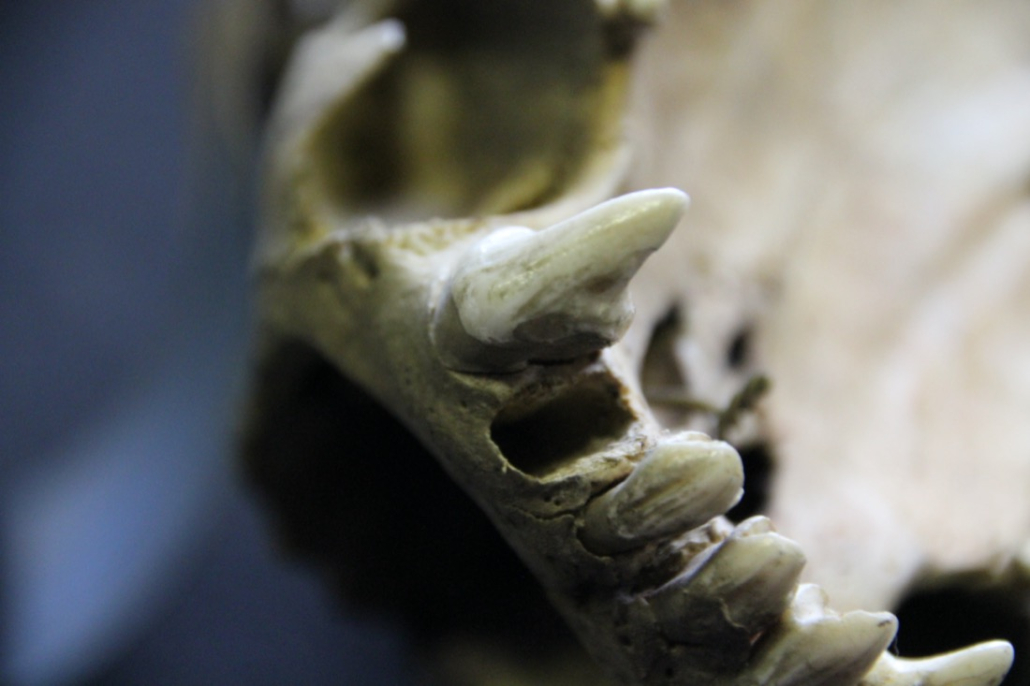
Teeth have a lower risk of contamination with modern or environmental DNA compared to other bones in the body, especially porous ones, since they are like a sealed box that preserves DNA from extreme environmental conditions. Therefore, we primarily use single-rooted polar bear teeth (incisors and premolars) for DNA extraction.
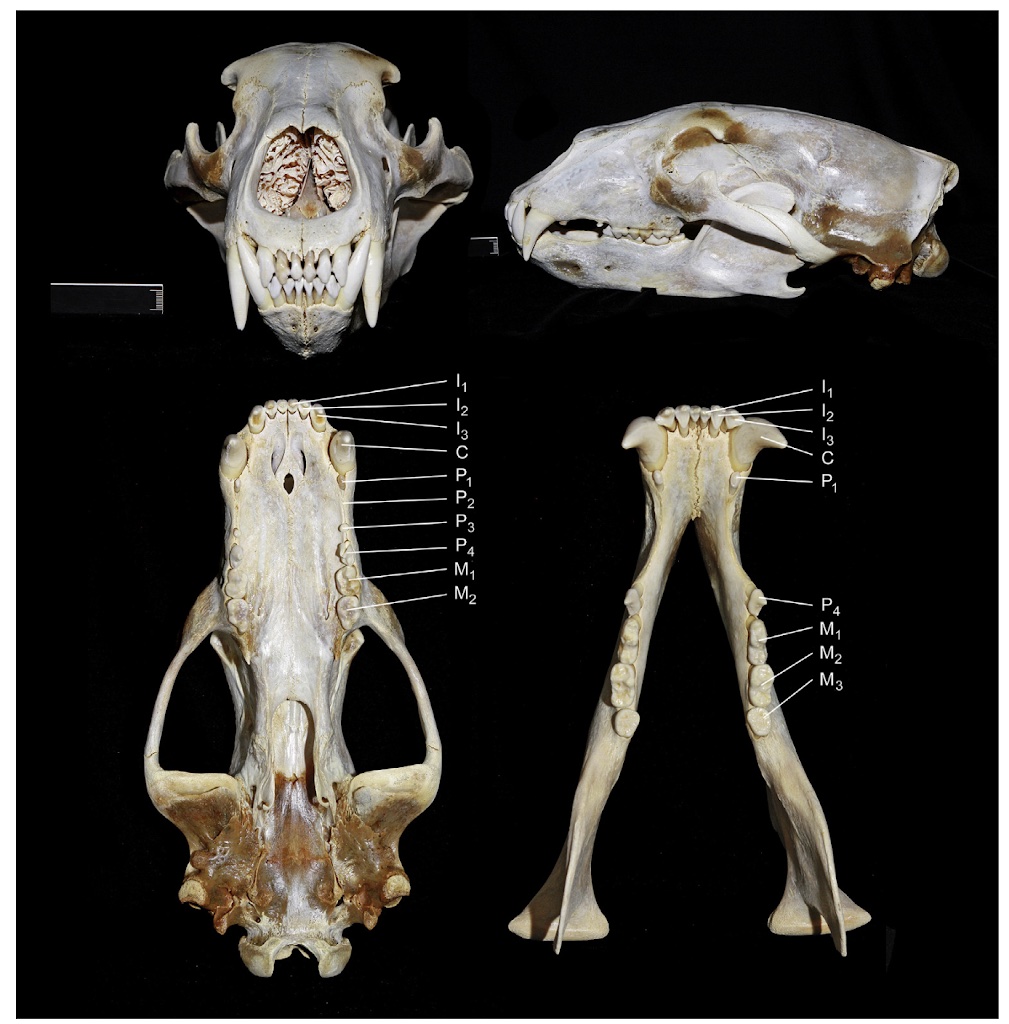 © Winer, J N et al. “Dental and Temporomandibular Joint Pathology of the Polar Bear (Ursus maritimus).” Journal of comparative pathology vol. 155,2-3 (2016): 231-241. doi:10.1016/j.jcpa.2016.07.004
© Winer, J N et al. “Dental and Temporomandibular Joint Pathology of the Polar Bear (Ursus maritimus).” Journal of comparative pathology vol. 155,2-3 (2016): 231-241. doi:10.1016/j.jcpa.2016.07.004
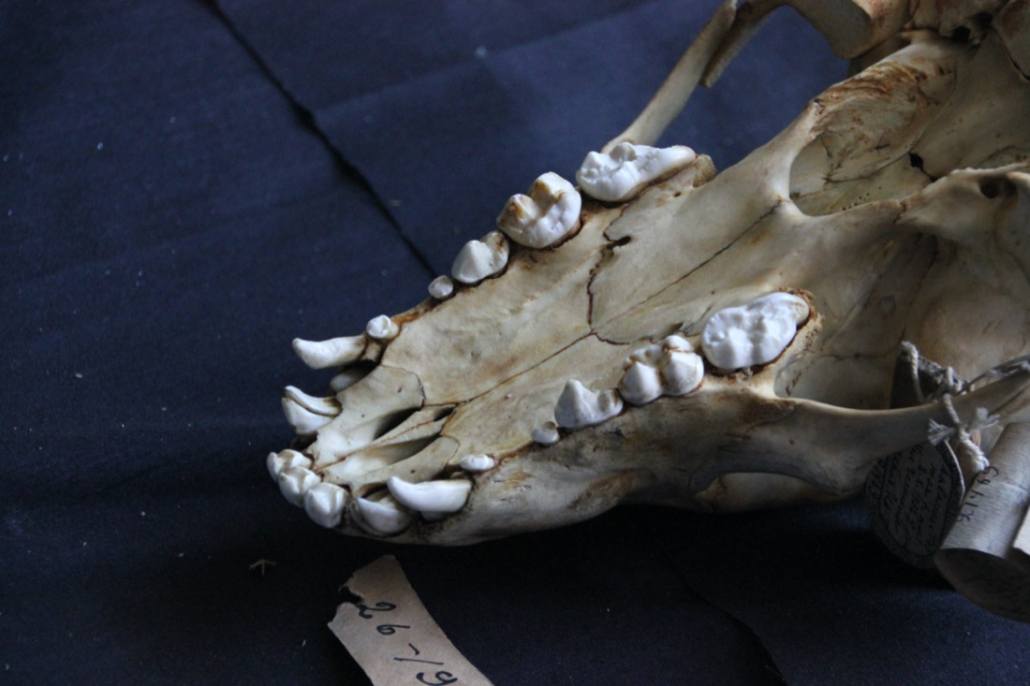

The sampling process is straightforward: the teeth are carefully extracted using dental instruments (many thanks to “MARKA” dental clinic (https://klinikamarka.ru/) for supplying us with tools) and barcoded. Next, the skull and the tooth are photographed to collect meta-information on the collection date, collector, geo-location, etc.



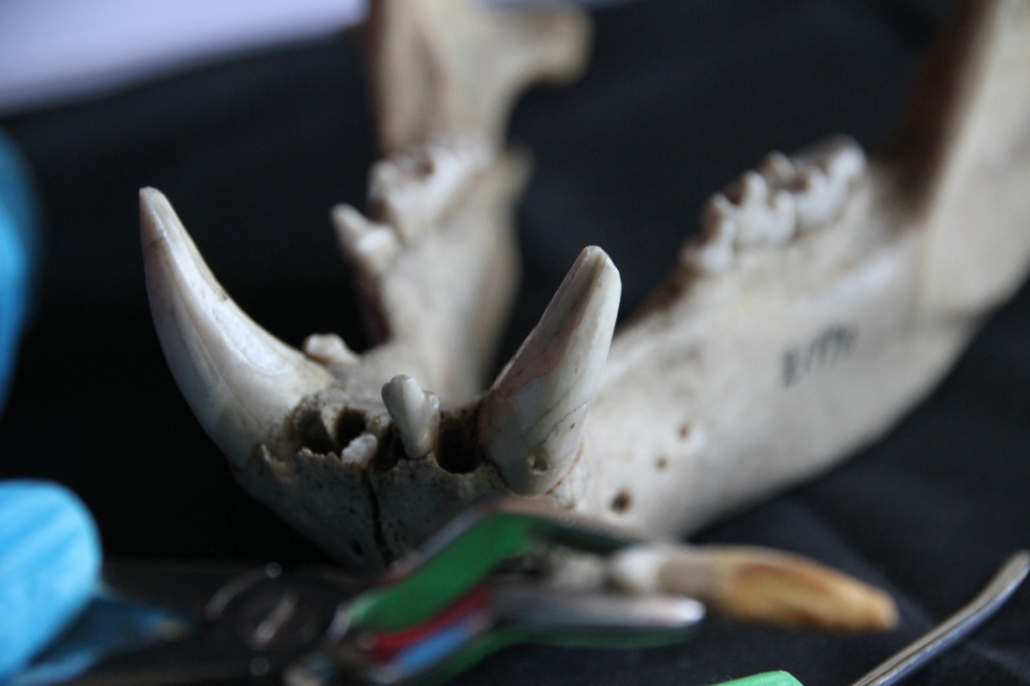

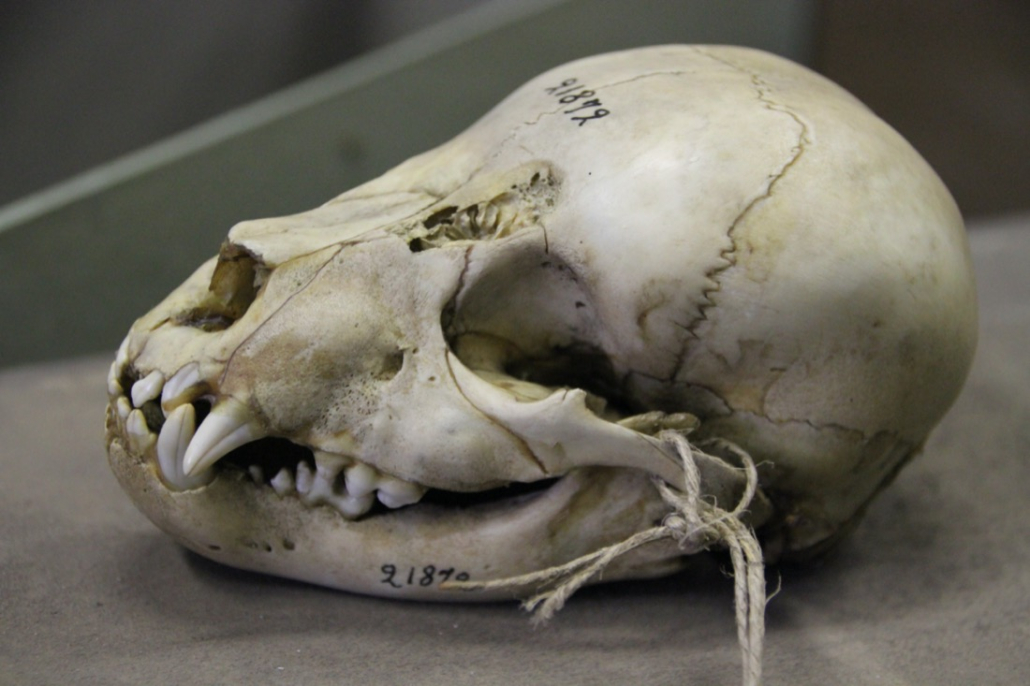

The collection contains over 450 polar bear skulls, including those found in indigenous peoples’ sacred sites.


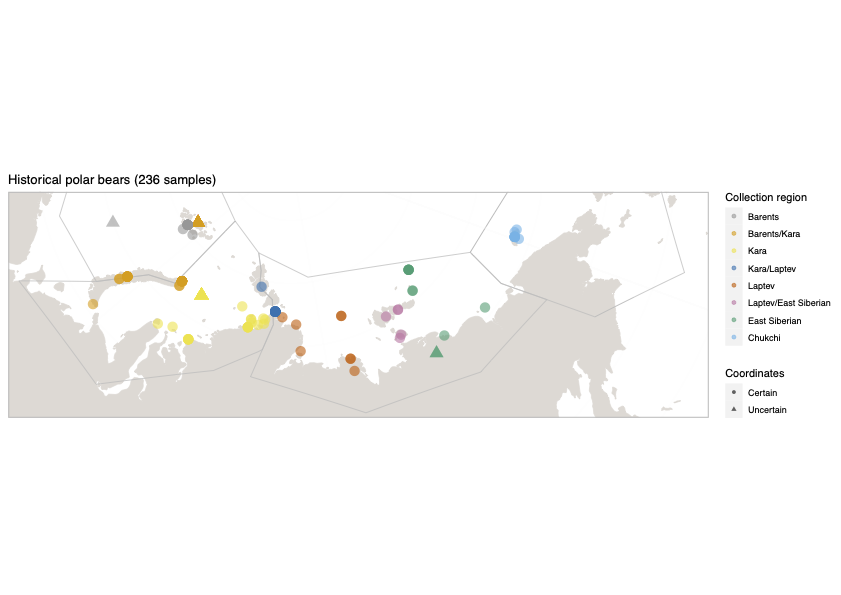
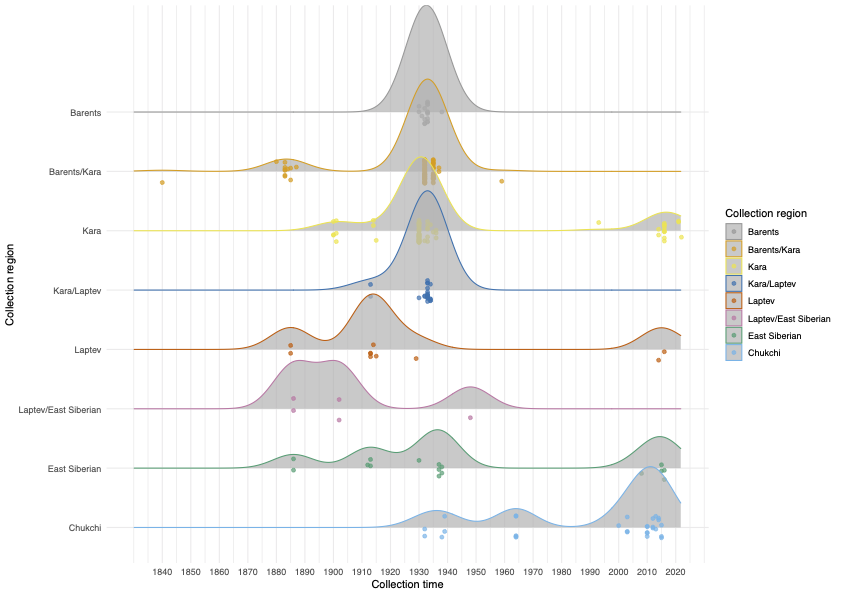
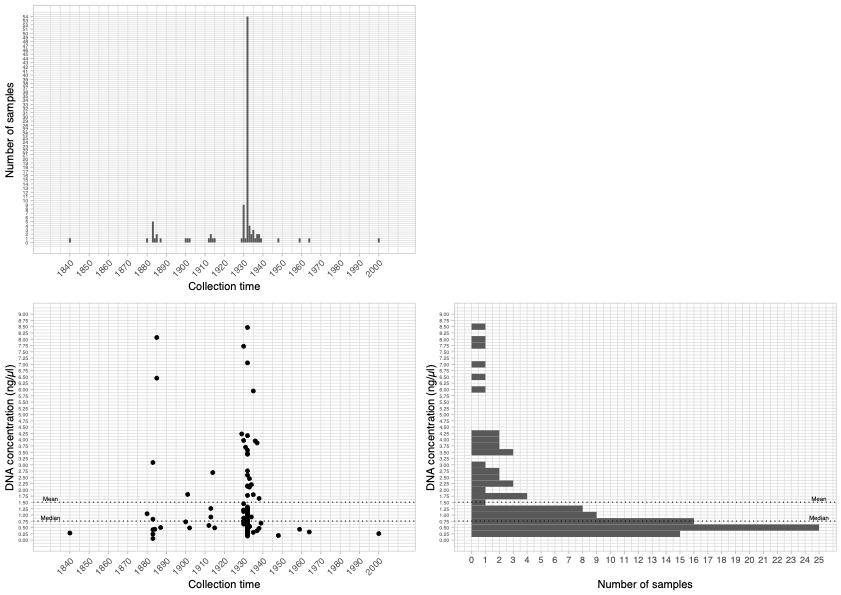
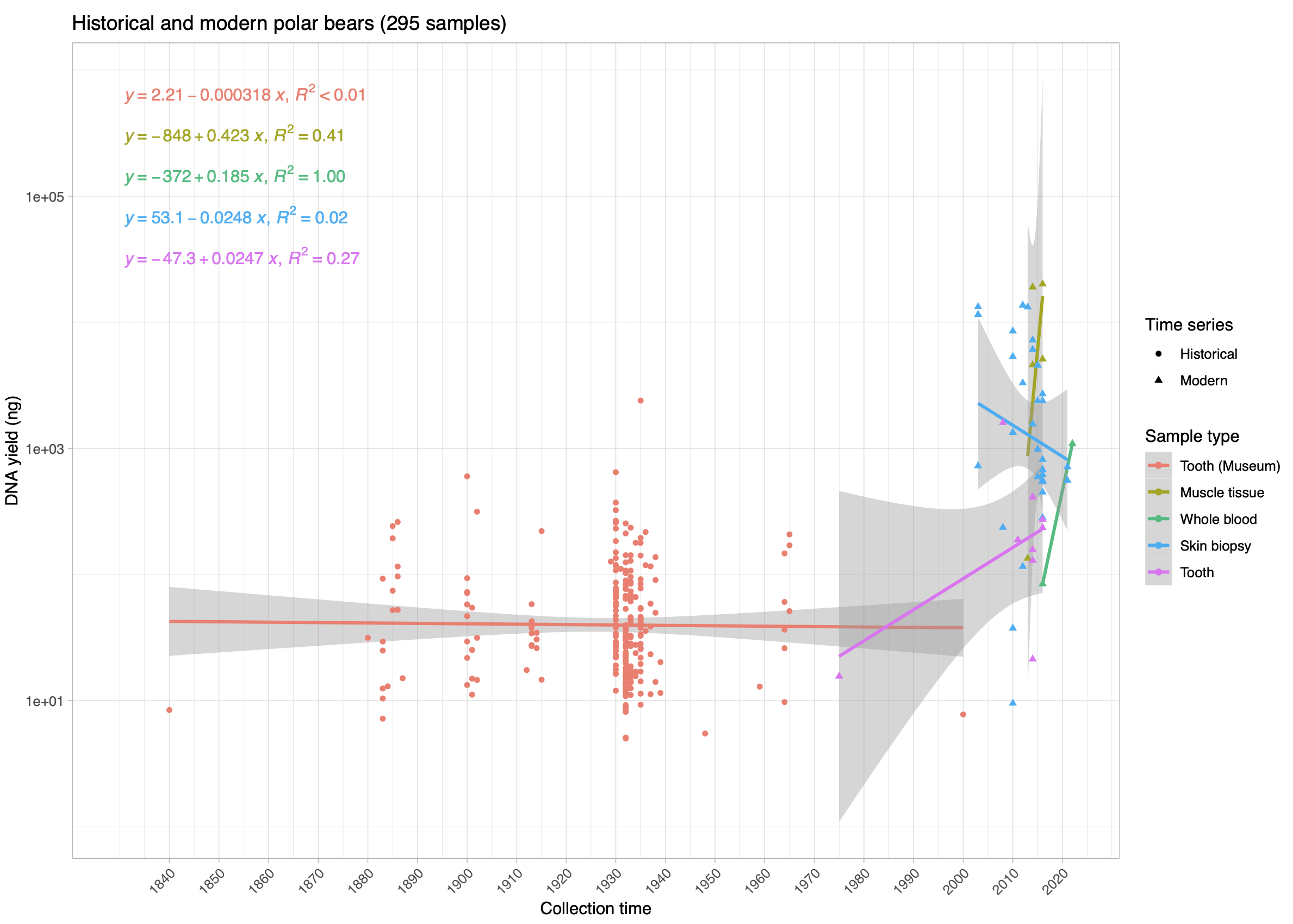
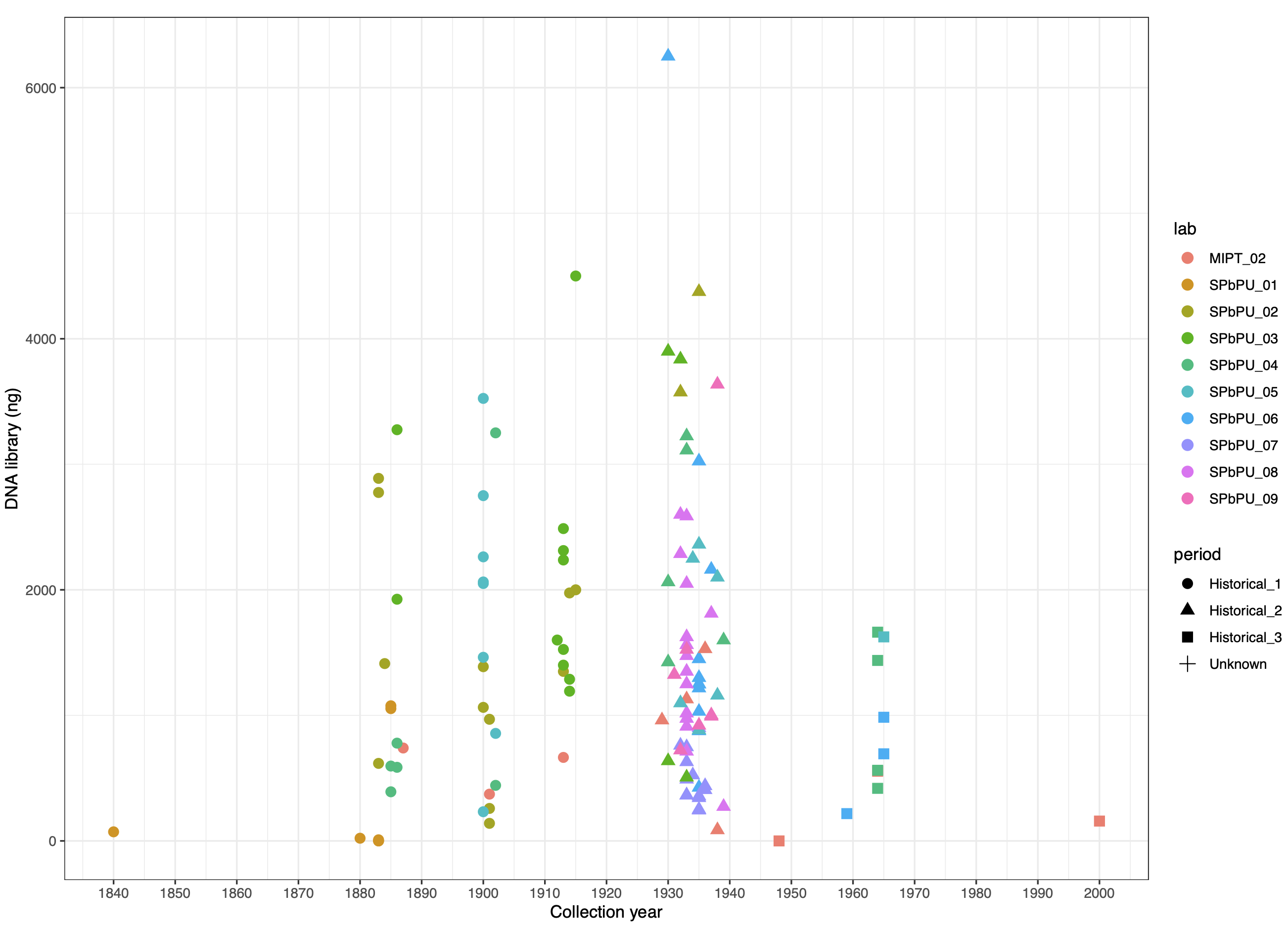
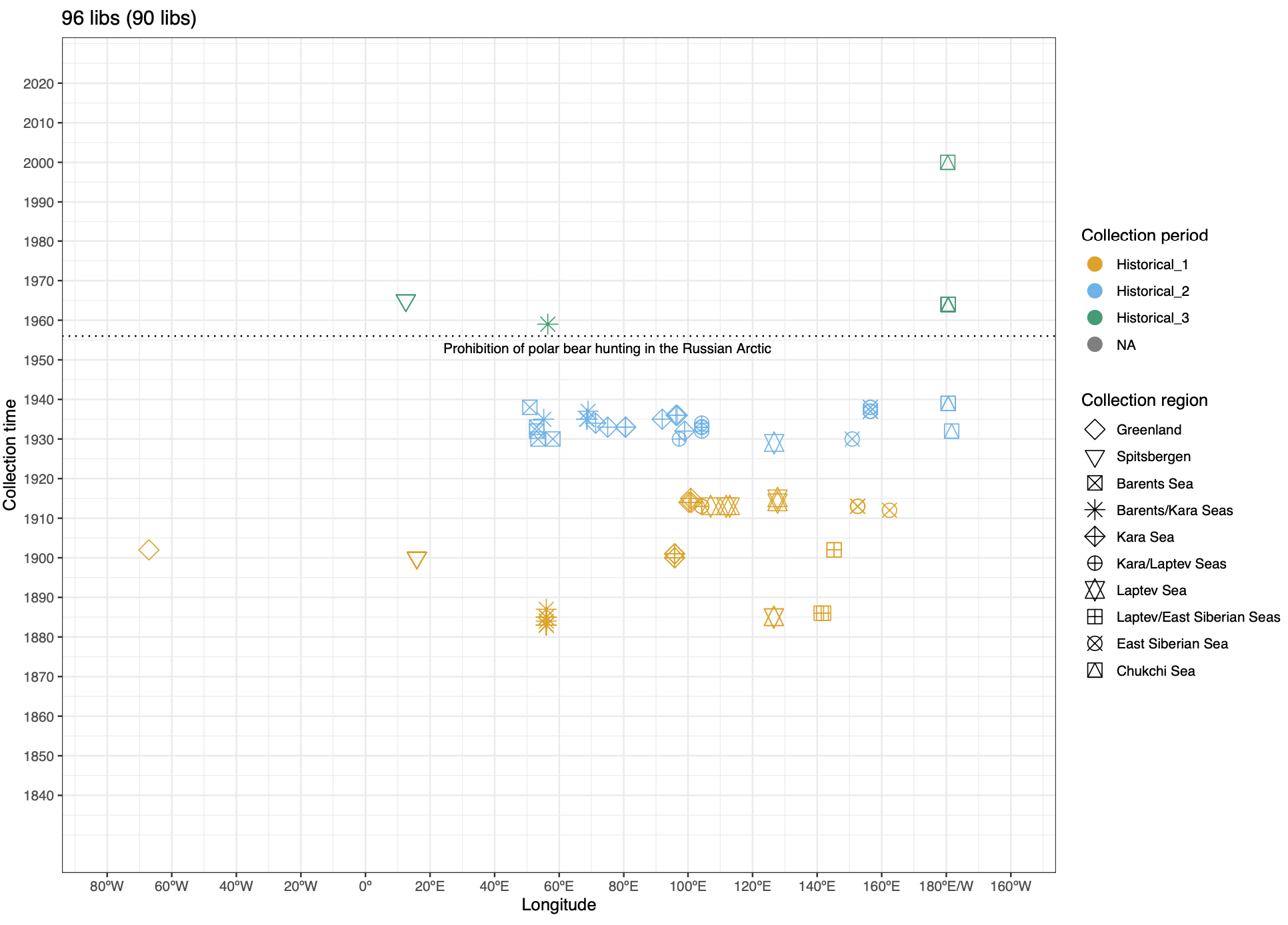
A significant number of specimens are hunting trophies obtained in the Russian Arctic between 1930-1940 when polar bear hunting was legal. To avoid unnecessary redundancy, we selected a representative subset of specimens for further work. The maps and graphs below provide an overview of the sample’s geographical distribution and age.
2022
November - December
The analysis of DNA from skeletal tissues and bones has revolutionized many fields of study, ranging from ancient DNA research to forensics and medical science. However, extracting small quantities of highly degraded DNA from such material is not an easy task. Short DNA fragments are challenging to isolate from other organic molecules, such as humic acids, which can inhibit enzymatic DNA manipulations that need to be performed prior to sequencing. In addition, when DNA is extracted and analysed from ancient samples, the fragments are often short and may contain lesions that impede the sequencing process and lead to the incorporation of incorrect nucleotides during DNA replication. Accurate DNA extraction is, therefore, one of the cornerstones of successful experimentation.
 © photo by M.Wallerstedt
© photo by M.Wallerstedt
 © photо by Reich Lab
© photо by Reich LabTypically, ancient or historical DNA is extracted in clean rooms, which are controlled environments with a low level of pollutants such as dust, airborne microbes, aerosol particles, and chemical vapours. These rooms are very expensive to build and difficult to operate because each specimen must be processed individually, and the entire room must be thoroughly cleaned and sanitized after each extraction. This is a time-consuming and tedious process.



Fortunately, there are alternative solutions that are just as effective. One such solution is the Historical Genetics Lab at the Moscow Institute of Physics and Technology, where an innovative approach to dealing with ancient DNA has been developed and is in active use. The experimental facility design reflects a recognition of the importance of a smooth process flow, with all experimental procedures being carried out in clean boxes with rubber arms.




These boxes are much easier to manage and operate and are cheaper to build, allowing for the implementation of DNA processing “conveyors” and a significant increase in throughput. With the current version of the extraction protocol and facility configuration, a hardworking, motivated researcher can manage DNA extractions from six samples in one day while successfully avoiding cross-contamination and achieving high-quality DNA extraction.
2022
December
The sample processing routine begins with the thorough scrubbing of teeth extracted from museum specimens, which were glued to the jaws using PVA. Before any molecular biology work can be done, the teeth must be cleaned from dust and chemicals, treated with UV radiation, and sterilized to ensure the accuracy of the results. ed, treated with UV radiation, and sterilized to ensure the accuracy of the results. The teeth are then cut into smaller fragments, as they are often quite large and sufficient DNA could be extracted from small bits. These pieces are then milled into a fine powder, from which DNA is extracted. For a more detailed step-by-step guide on this process, see below.
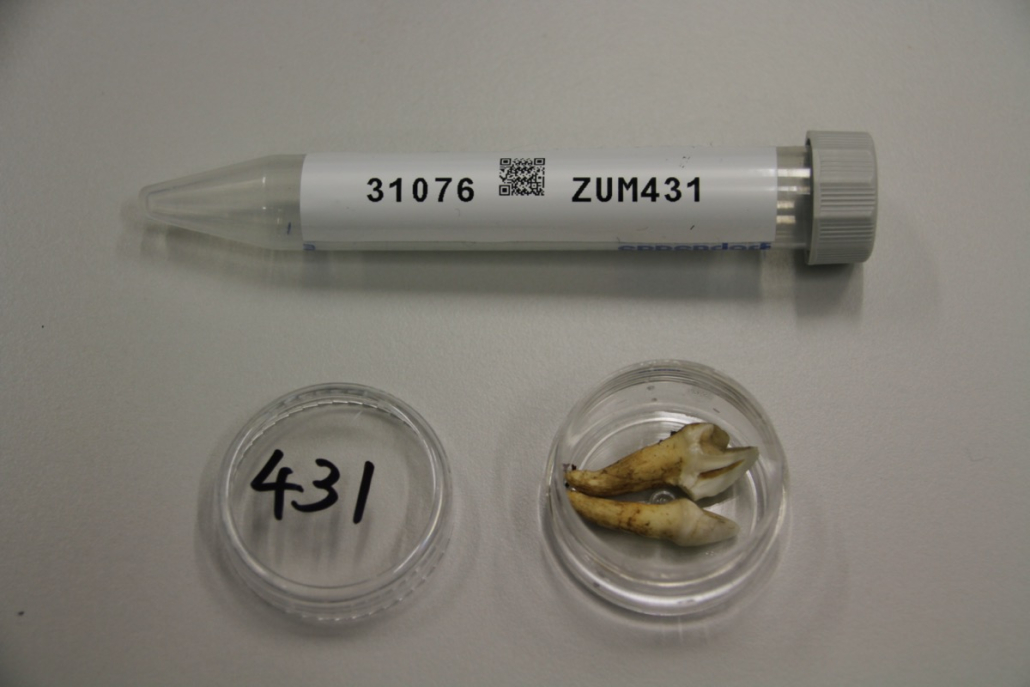

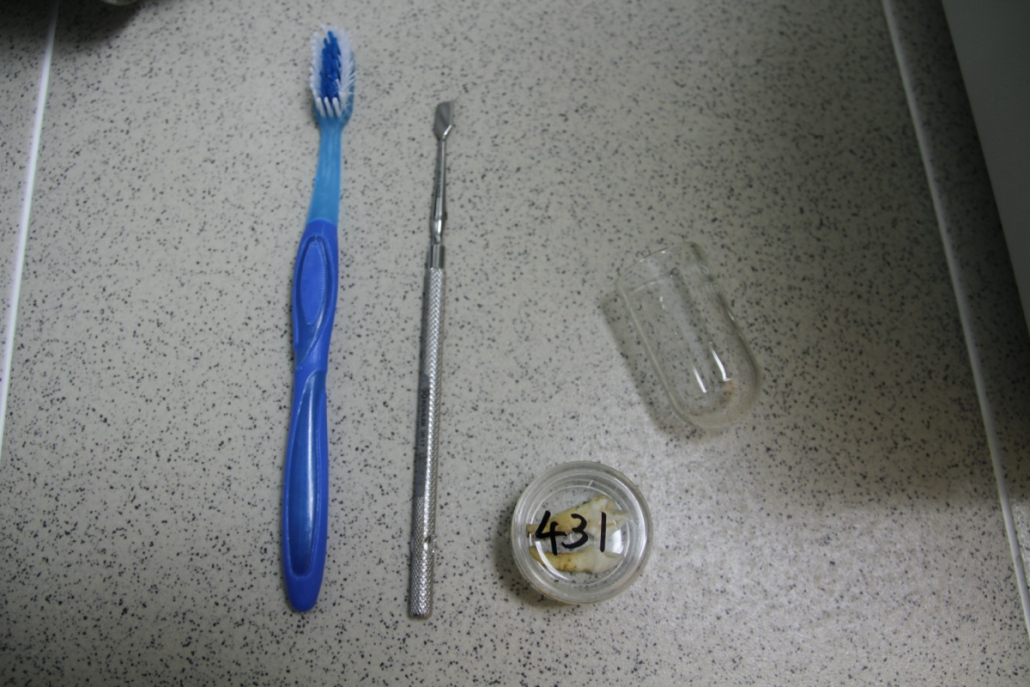

The teeth are thoroughly cleaned in a sonicated bath and cut using dental drills, the fragments are milled into fine powder, and the powder is then weighed and transferred into storage tubes for further use.




The DNA extraction and subsequent sequencing library construction are also performed in clean boxes.







Since the inception of our project, we have successfully extracted over 100 samples of various origins and ages. As a preliminary step to estimate the extent of bacterial and other types of contamination, we plan to conduct a shallow whole genome sequencing experiment on two of these samples, analysing their entire genome. Moreover, we are eagerly anticipating the results of low concentration DNA samples, where enrichment was not performed, to ascertain whether any valuable insights can be gained from them.
2023
January-February
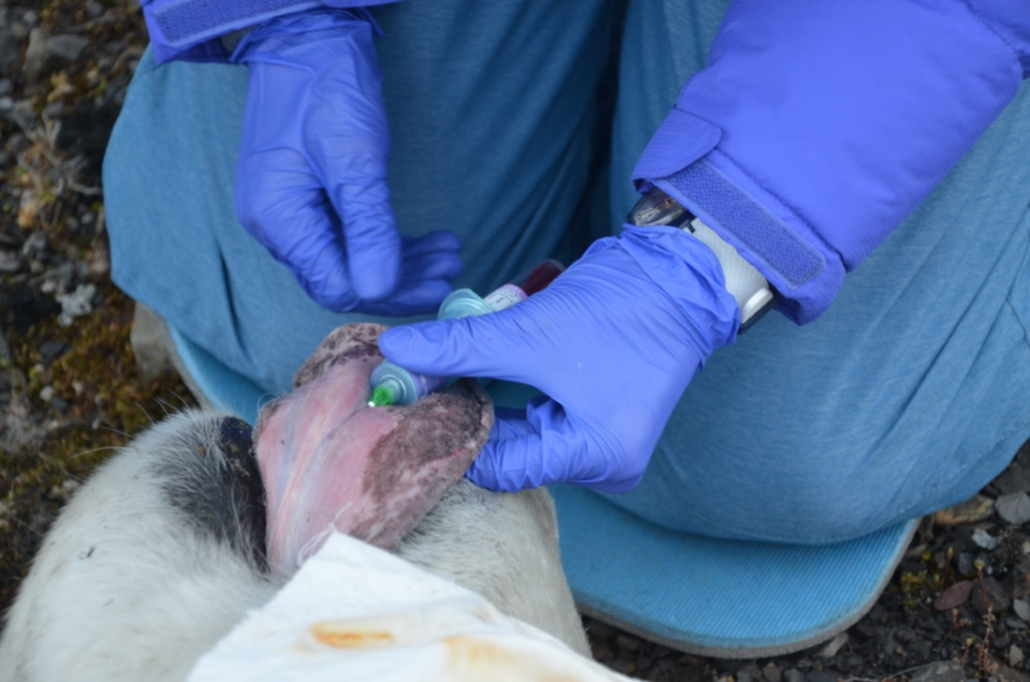
The project revolves around comparative analyses of historical and modern polar bear DNA, and the hyRAD experimental protocol requires modern DNA for enrichment via a hybridization-capture approach. But for those who do not reside in Vankarem, Dikson, or Churchill, Canada, obtaining modern DNA can be challenging. Fortunately, our collaborators at the Marine Mammals Research Expeditions Company (mmrec.ru) have done the hard work for us by providing polar bear teeth and tissue samples (blood, muscle, skin) to use in our project. The tissue samples were primarily collected from live animals, although some were obtained from remains.
 © mmrec.ru
© mmrec.ru2023
March
Hooray! Hooray! After months of hard work, the first sequencing results have arrived!
The purpose of this sequencing run was a) to assess if the library construction protocol works in our hands, to estimate the extent of contamination of our samples with various pollutants, and, finally, c) to check if the samples with low DNA concentration would yield any meaningful results in the absence of enrichment (i.e., with shallow whole genome sequencing).
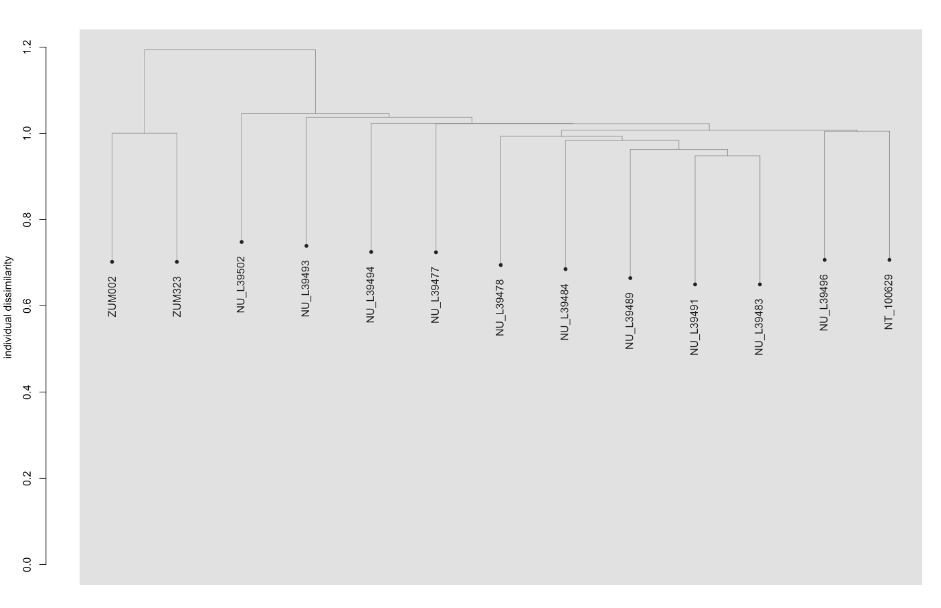
We chose to sequence two samples, ZUM002 and ZUM323, for our comparative analysis of historical and modern polar bear DNA. ZUM002 is one of the oldest samples in our collection, dating back to 1883 when it was collected by D.R. Grinevetsky on Novaya Zemlya Island. ZUM323 was collected in 1930 by hunters from the “New Export” Agency in the Kara Sea. After DNA extraction and library preparation, we quantified the DNA using Qubit and found that ZUM002 had a low concentration with readings of 0.063 and 0.94 ng/µl, while ZUM323 had a higher concentration with readings of 1.2 and 2.615 ng/µl. The values above show concentrations after DNA extraction and DNA library prep, respectively.
When we analyzed the fragment distribution of the DNA libraries using the Agilent Bioanalyzer instrument, we observed that ZUM002 had a small amount of DNA with a peak around 200 bp and many short dimers (< 100 bp). In contrast, ZUM323 had a larger amount of DNA with relatively longer fragments (> 300 bp).
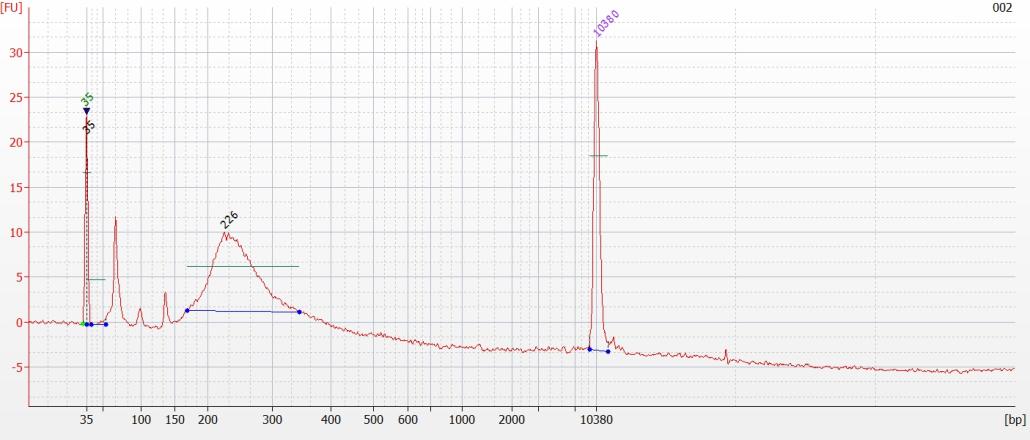
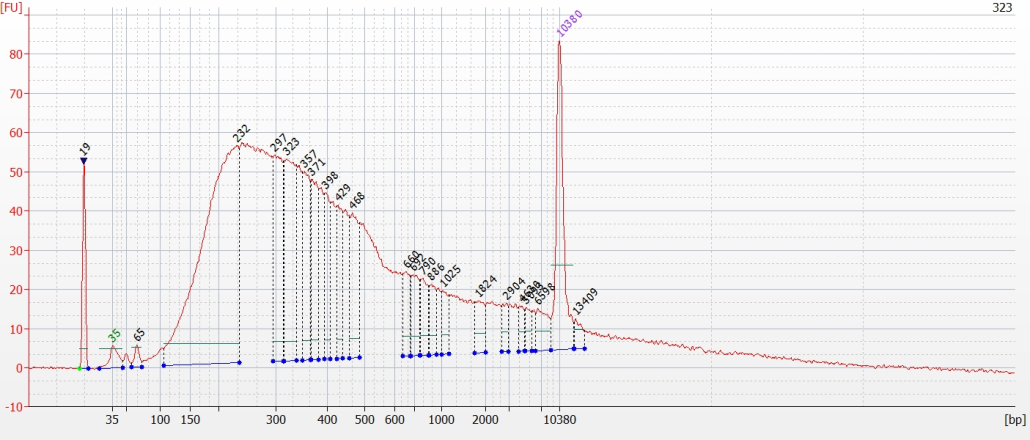
To align the sequencing reads to the polar bear genome (UrsMar v1.0), we filtered out low-quality reads and found that 62.2% of ZUM002 sequencing fragments and 98.6% of ZUM323 reads were aligned to the genome. We covered 9% and 21% of the polar bear genome for ZUM002 and ZUM323, respectively, with a coverage of 3x or above. This indicates that the bacterial contamination of our DNA samples is relatively low, and we hope to achieve even better results with the enrichment protocol.
We compared our samples to 10 randomly chosen Canadian polar bear samples published in (a reference), and we were delighted to find a clear distinction between our samples and the published data. Although this is an early result, it is an exciting one.
2023
April
Preserved samples found in museums provide an abundant and invaluable source of specimens for research. Historical samples, in particular, offer tremendous research potential and have led to the development of several protocols aimed at reliably sequencing reduced-genome subsets in non-model organisms. One such protocol is the hyRAD (Hybridization restriction-site associated DNA) protocol, which was designed specifically for use on degraded samples like archival DNA from museum collections.



In short, the protocol involves using DNA fragments generated through double digestion RAD protocol (ddRAD applied to fresh samples) as hybridization-capture probes to enrich shotgun libraries made from historical specimens in the fragments of interest. This approach allows for the acquisition of large sets of homologous loci from museum specimens, without requiring any prior genome information.
However, the experimental protocol can be tricky, and there is potential for many things to go wrong. We are following the approach described by Suchan et al. (see GitHub and paper), but we have also made several modifications, which will be explained in detail later on.
2023
May – June
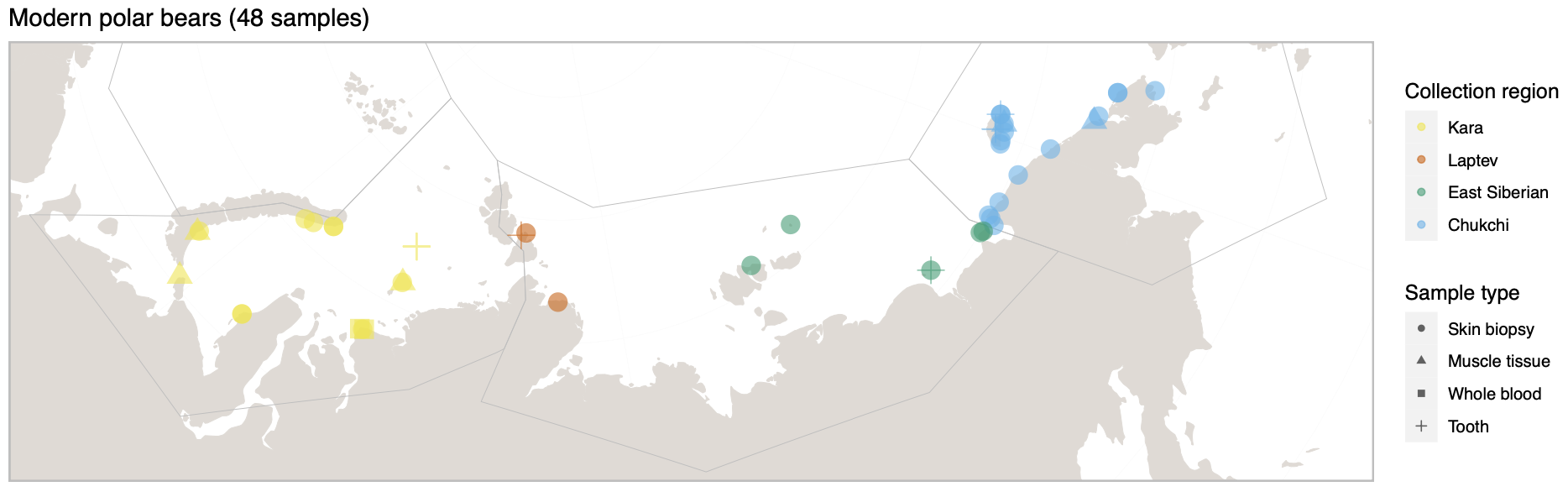
Milestone TitleWe have successfully completed the DNA extraction of all 252 historical polar bear samples from museum teeth specimens. Additionally, DNA extraction of all 48 modern polar bear samples from muscle tissues, whole blood, skin biopsies, and teeth collected from a diverse range of locations in the Russian Arctic region, spanning from Novaya Zemlya to the Chukchi Peninsula, between 1975 and 2022, provided by Dr. Andrei Boltunov (Marine Mammal Research and Expedition Centre, Moscow), has been completed.
2023
August
Quality Control (QC) of the extracted DNA from all historical and modern samples was performed by assessing DNA concentration, DNA degradation level, and DNA purity.

2023
July – October
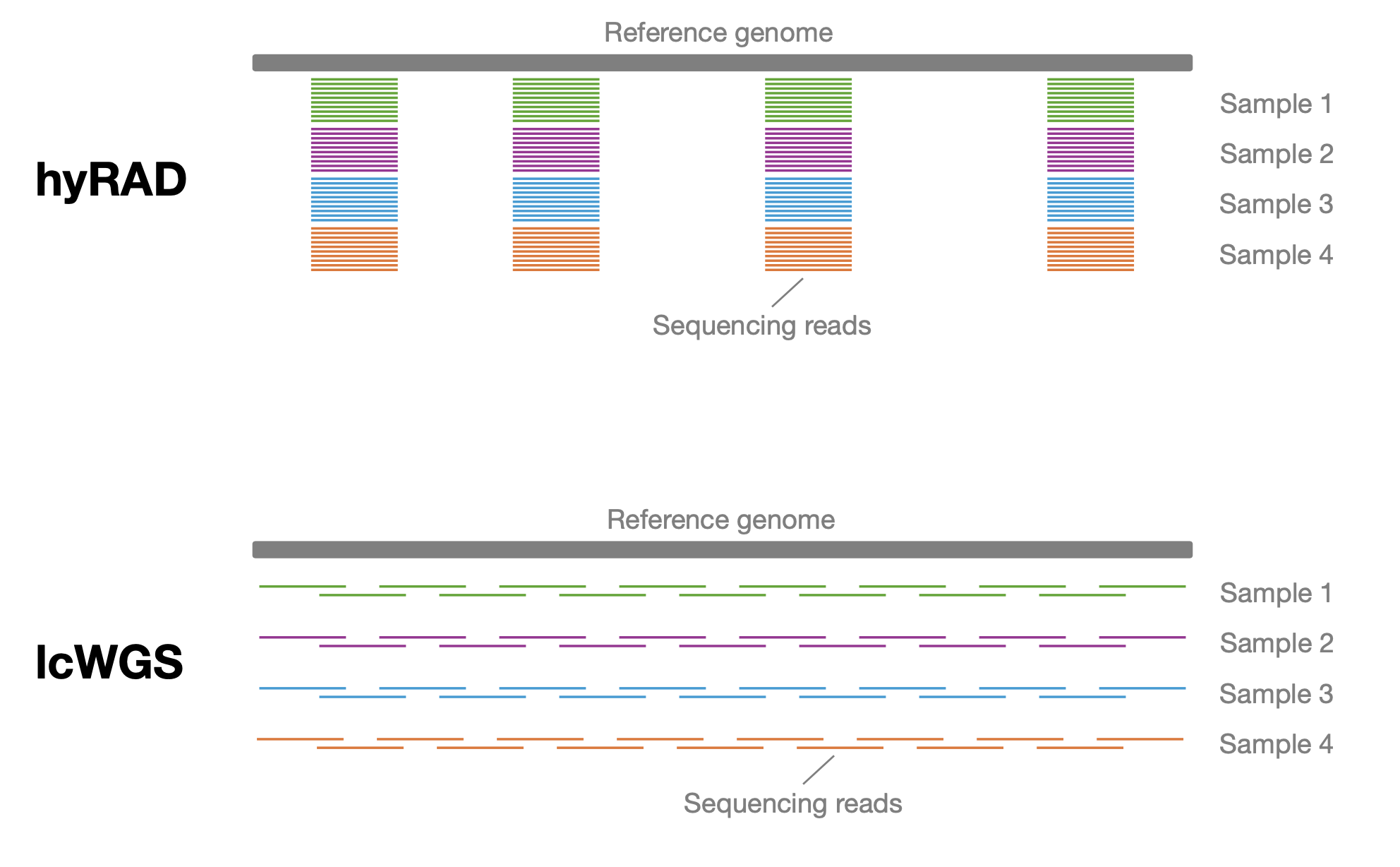
Hybridization RAD (hyRAD) is a technique that involves using DNA fragments generated from fresh samples through the double digestion RAD (ddRAD) protocol as hybridization capture probes to enrich shotgun libraries specifically for fragments of interest (Suchan et al. 2016). hyRAD experiment is composed of three different parts of experiment, (1) generation of ddRAD probes for hybridization capture using fresh modern polar bear samples, (2) DNA library prep of historical polar bear samples, and (3) hybridization capture of historical polar bear DNA libraries and modern polar bear ddRAD probes. Trial and error were attempted for setting up reaction conditions for the hyRAD experiment, but it did not work well for making ddRAD probes, so we decided to abandon this experiment.
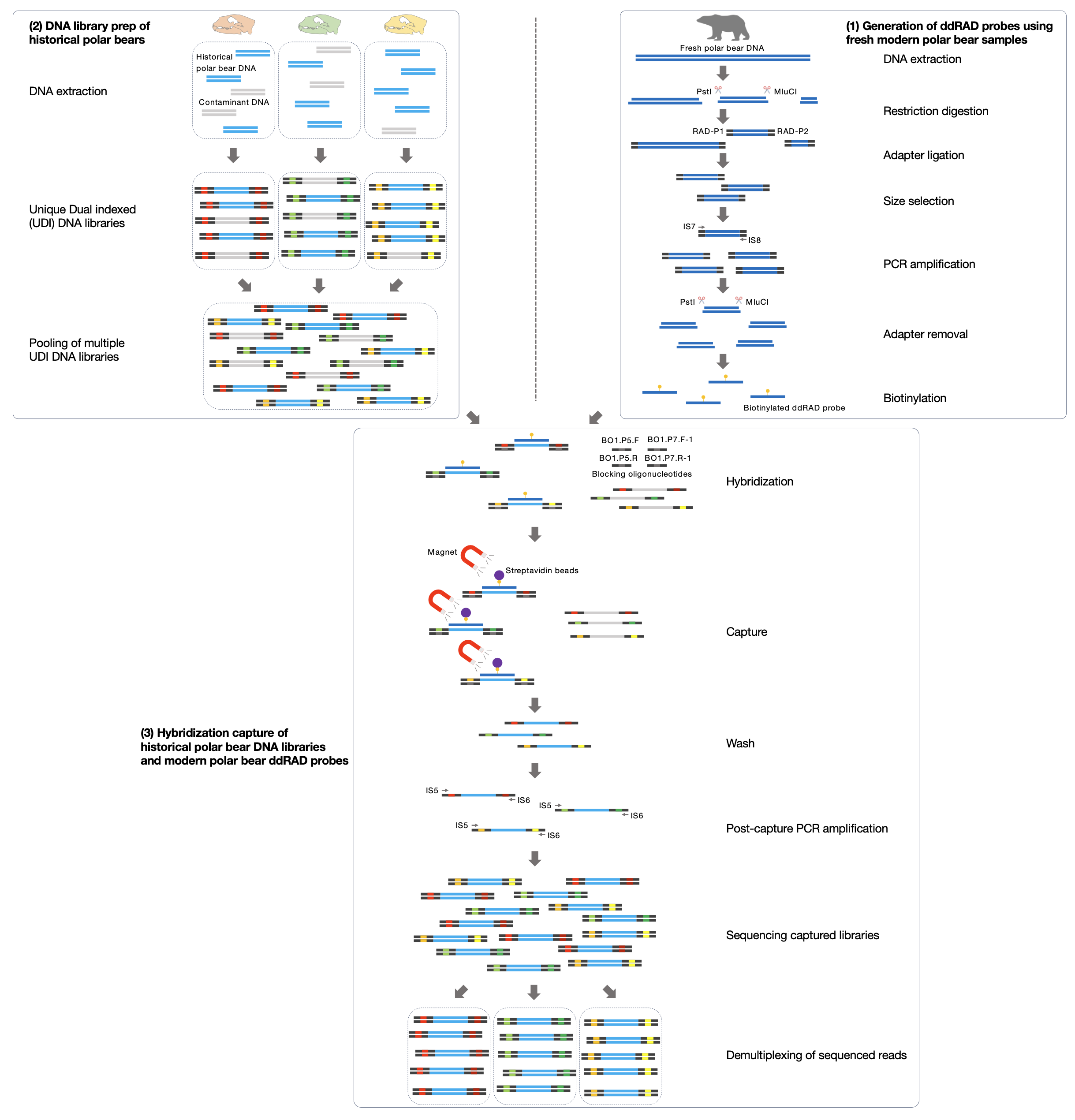
Alternative approach of hyRAD, we applied shotgun sequencing to obtain low-coverage whole-genome sequences (lcWGS). Both hyRAD and lcWGS approaches have distinct advantages and limitations. The hyRAD method allows for sequencing many samples with high coverage of reads by targeting a specific set of loci through hybridization capture of DNA libraries. On the other hand, lcWGS enables the sequencing of a larger number of loci distributed across whole-genome sequences through shotgun sequencing of DNA libraries, however it is limited by low coverage of reads and the capacity to analyze a small number of samples.
2023
November – January (2024)
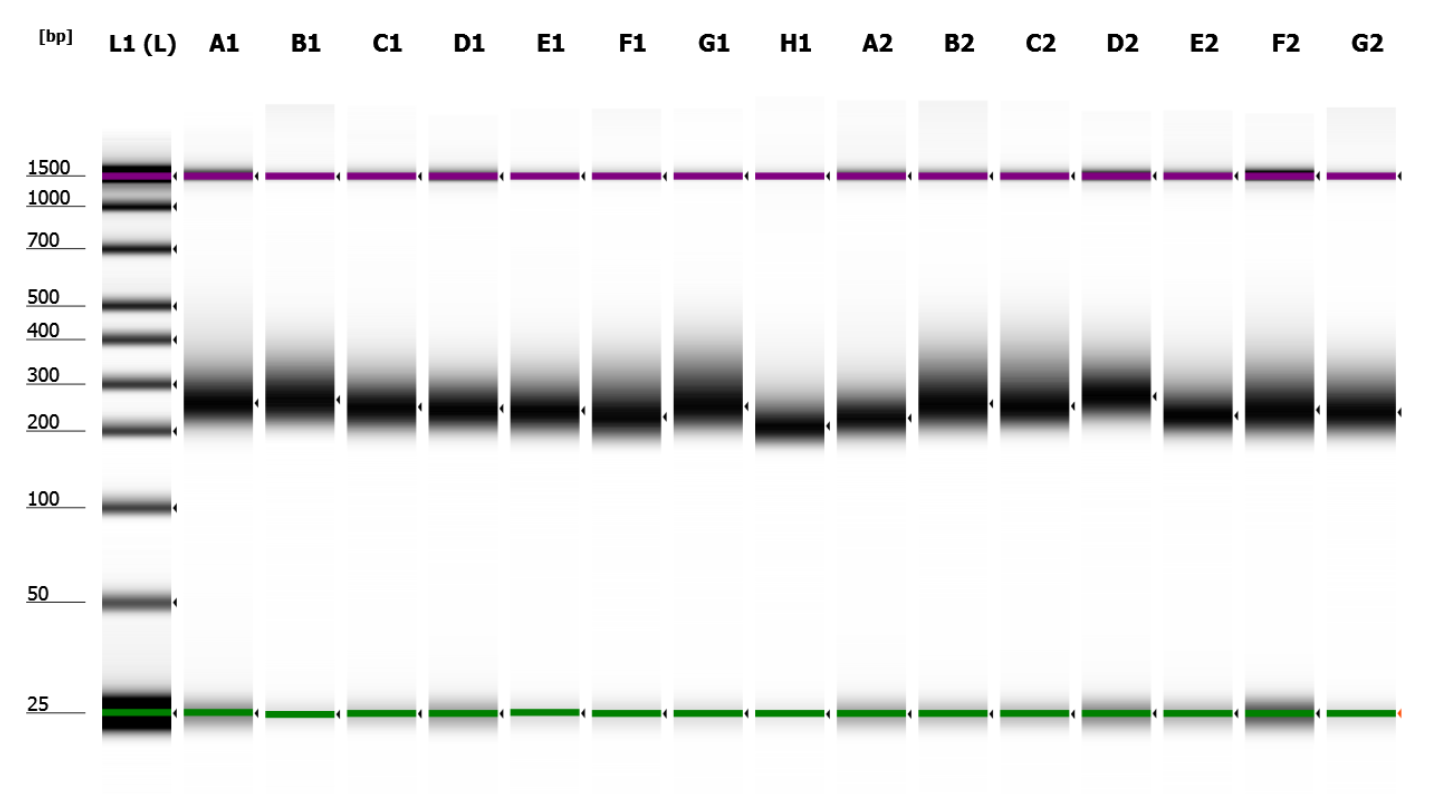
From a total of 252 extracted DNA samples of historical polar bears, 131 samples were selected for DNA library preparation based on the DNA yield of the prepared libraries, geographical distribution, and collection time of the samples. DNA concentration, fragment size, and quality of the prepared DNA libraries were assessed. Based on the obtained electropherogram, the DNA libraries, which showed the presence of primer dimers and/or adapter dimers, were purified using magnetic beads. After QC of the DNA libraries, 118 libraries were found to have sufficient quality and quantity for sequencing.
2024
January – February
Prepared DNA libraries of the historical polar bear samples were sent to Genetico in Moscow for sequencing.

Before sequencing, QC of the 118 DNA libraries was conducted again at Genetico. The QC process confirmed that all the libraries meet the required standards and are ready for sequencing.
Considering the fragment size, geographical distribution and collection time of samples, 96 DNA libraries for sequencing were selected.
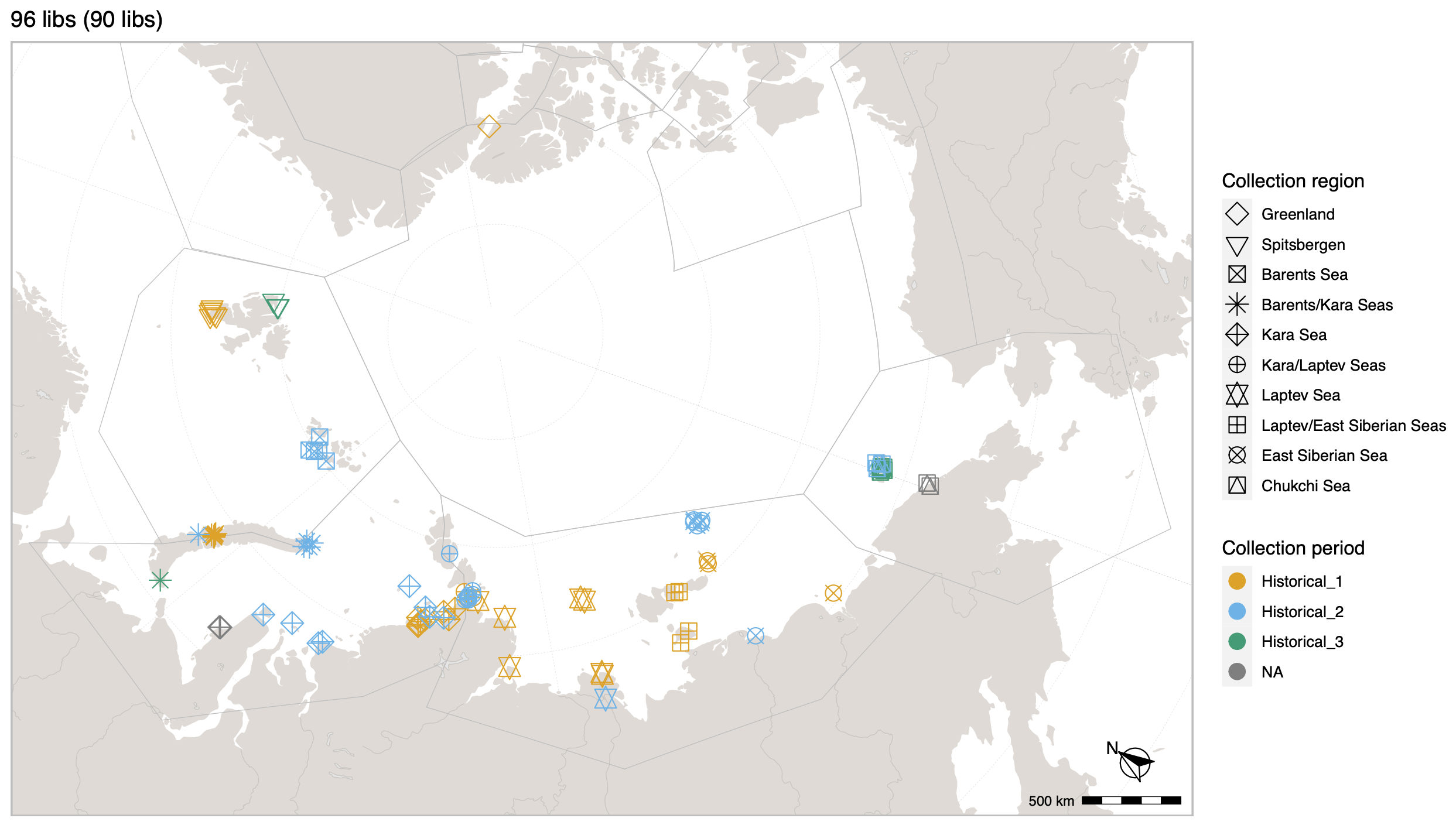
Sequencing runs of the 96 DNA libraries were performed using the NovaSeq 6000 platform (Illumina) with S2 chemistry (paired-end mode with 2×100 reads).
 © https://commons.wikimedia.org/wiki/File:NovaSeq_6000.jpg
© https://commons.wikimedia.org/wiki/File:NovaSeq_6000.jpg2024
March – May
Sequencing data were delivered from Genetico and we have started data analysis. We firstly inspected the contamination level of the obtained sequencing reads by checking the ratio of exogenous bacterial/archaeal DNA and endogenous polar bear DNA of the obtained sequencing reads using metagenomic tools. The contamination level and the remaining rate of polar bear DNA varied among samples, but older samples tended to have a higher rate of exogenous bacterial/archaeal DNA and a lower rate of endogenous polar bear DNA.
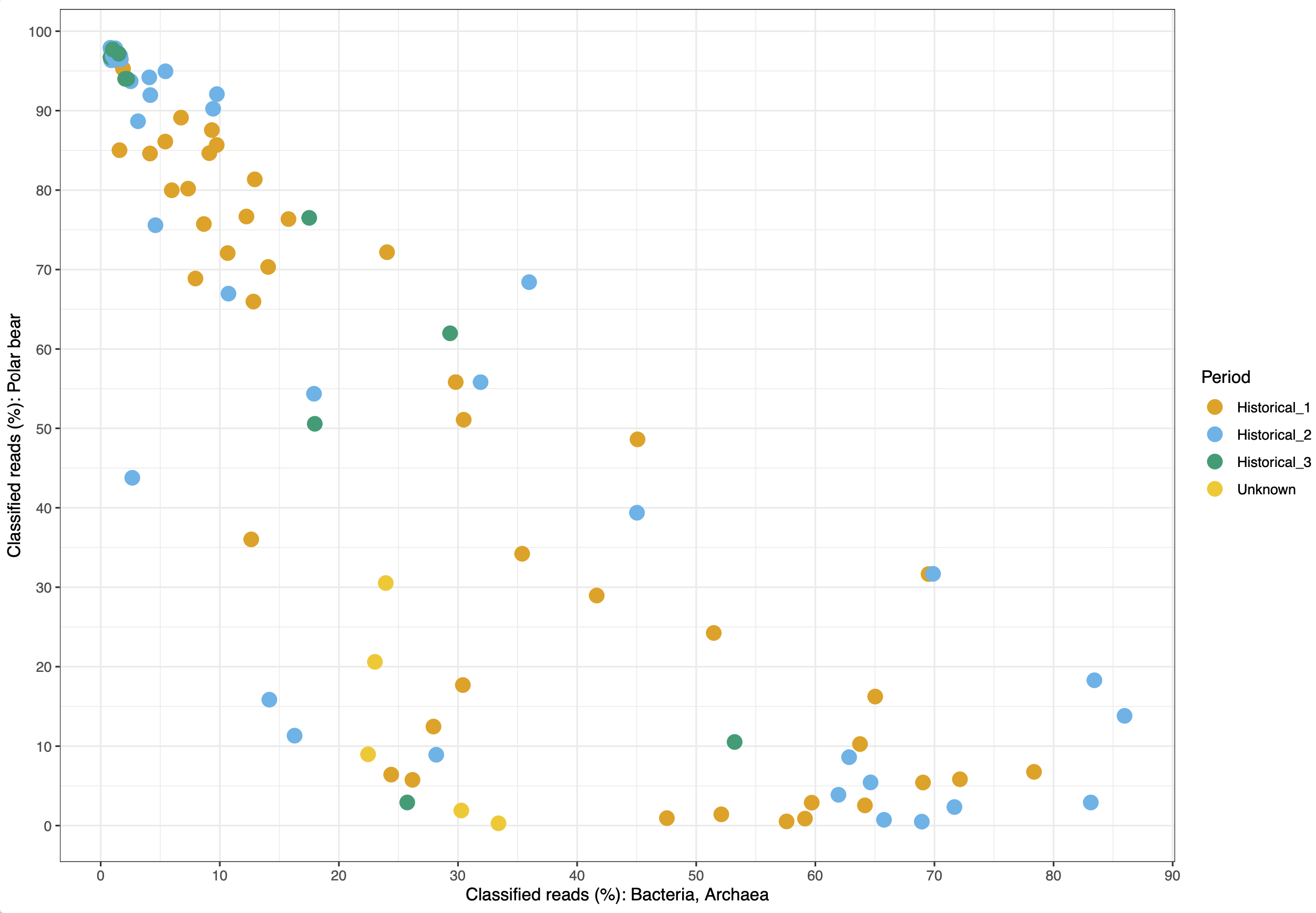
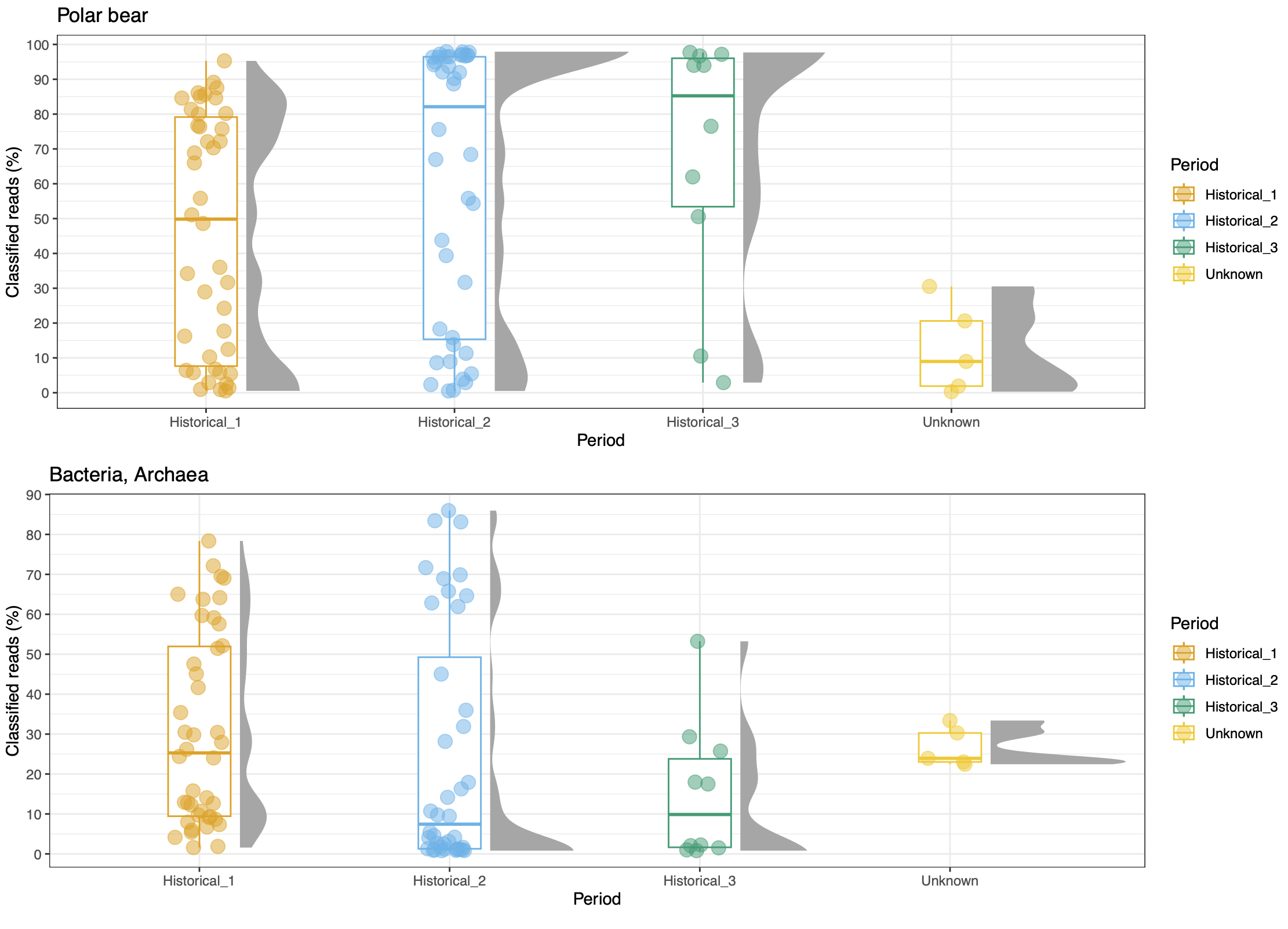
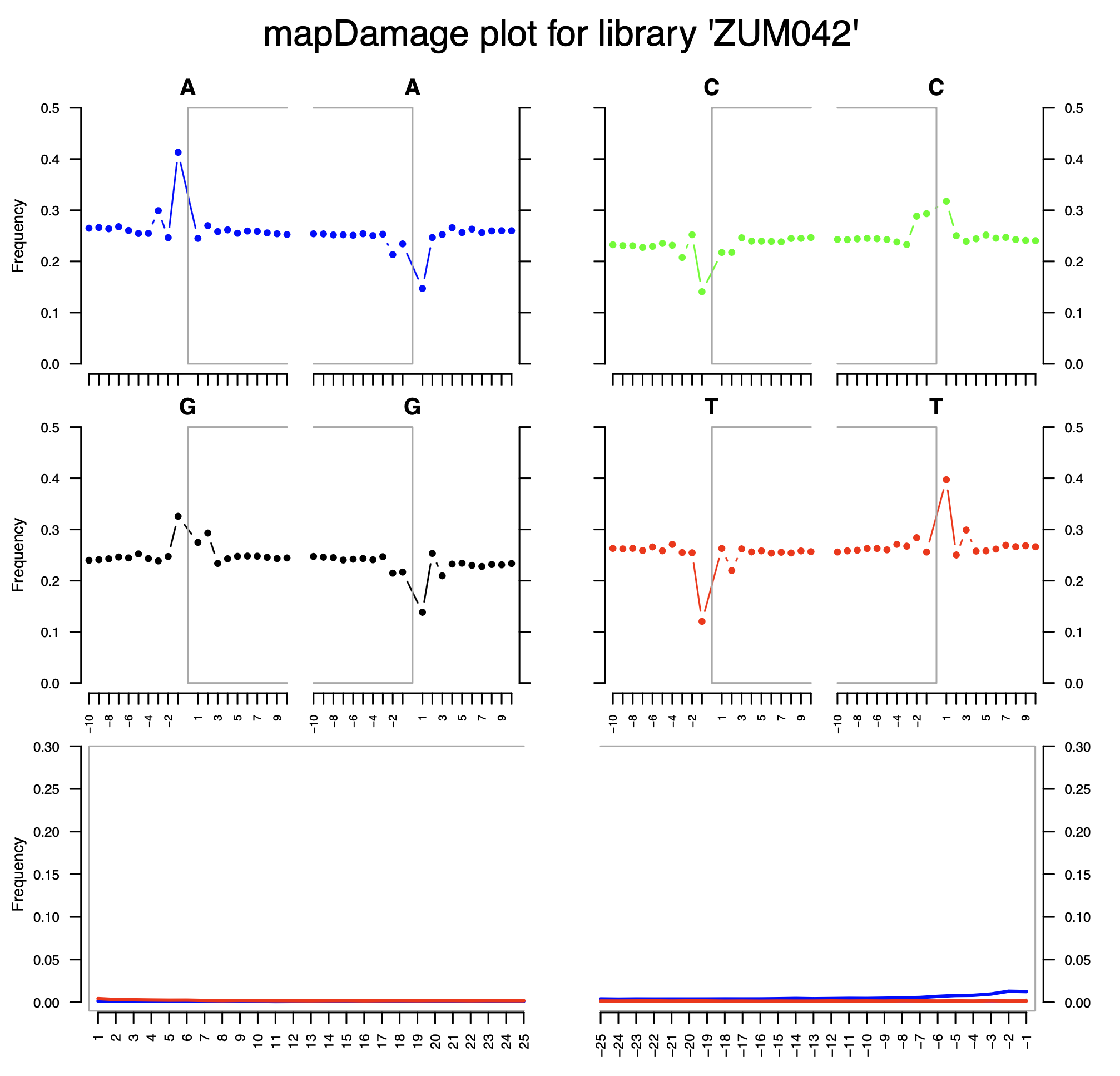
Then, we aligned the sequencing reads to the polar bear reference genome sequence. Mapping statistics including DNA degradation level were assessed after the alignment. Typical sequence patterns of post-mortem DNA damage observed in ancient DNA such as DNA fragmentation through depurination and signatures of post-mortem cytosine deamination (Orland et al. 2021) were observed in the obtained sequences.

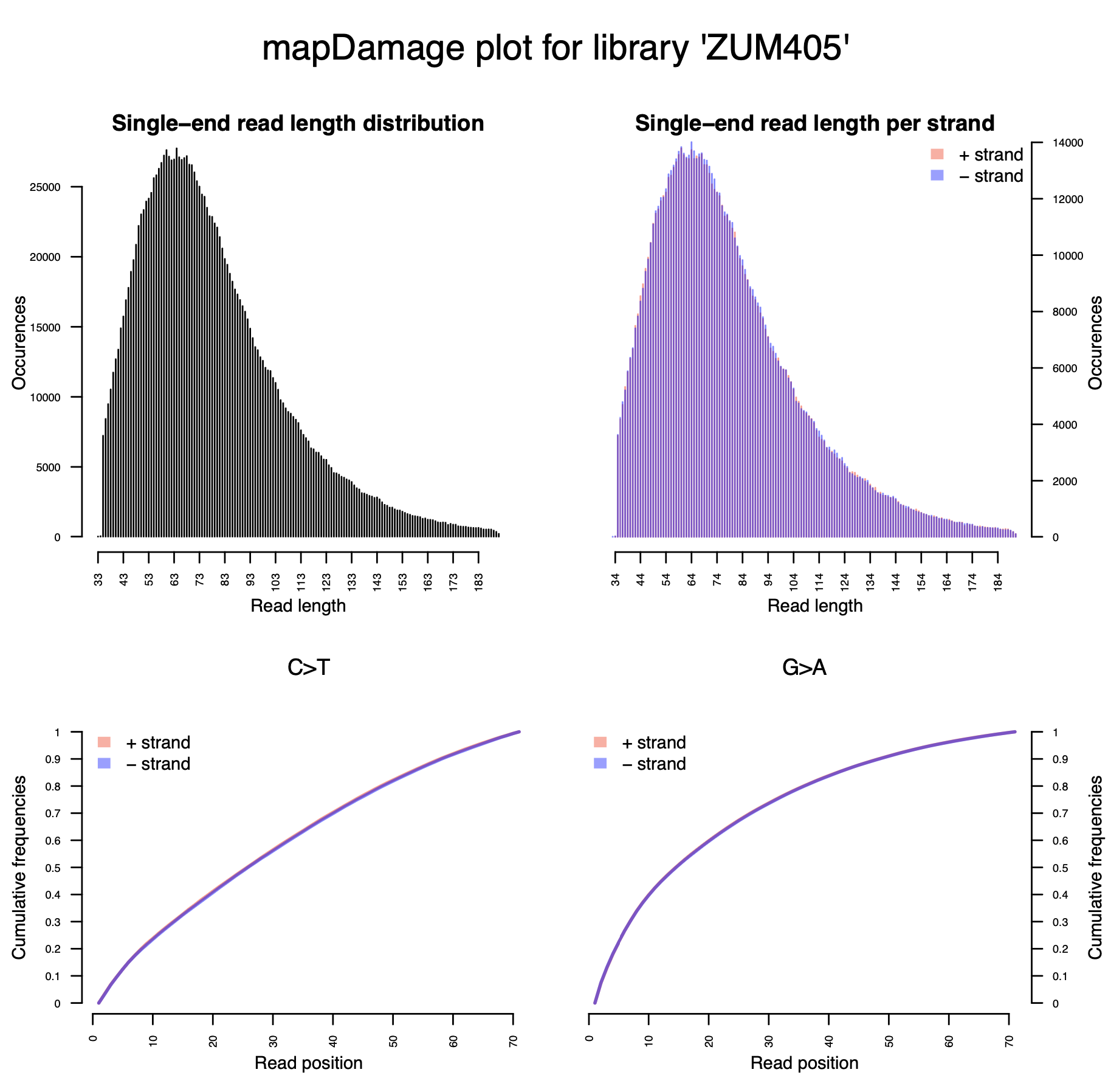
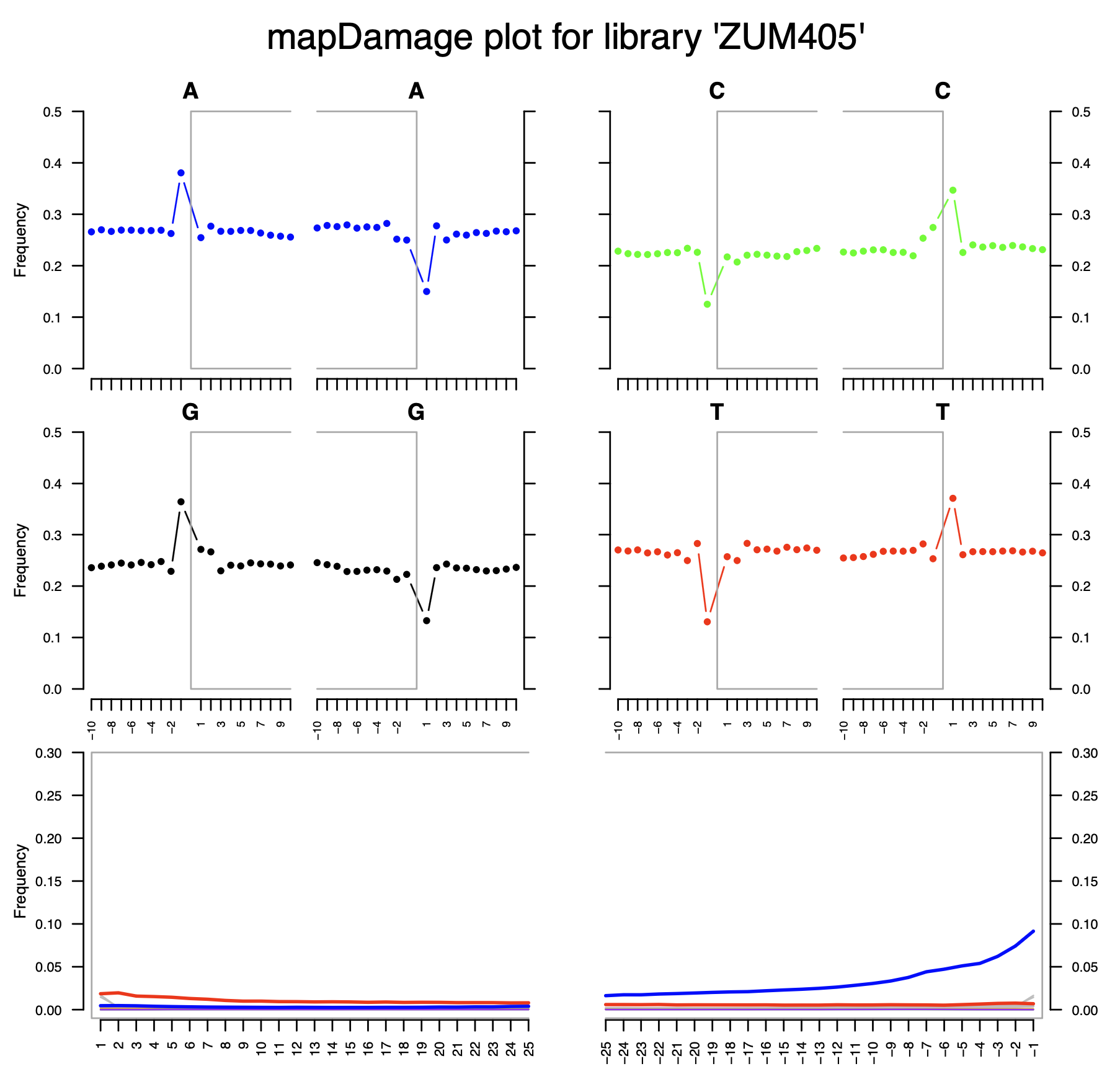
To reveal population structure and genetic diversity of polar bears in the past century, population genetic analyses are in progress based on the obtained alignments.